Posted On : Oct 07 , 2024
Posted By : CureSureMedico Team
Le rétinoblastome est un cancer rare mais potentiellement mortel de la rétine, un tissu sensible à la lumière situé à l'arrière de l'œil. L'un des signes les plus visibles de cette maladie est que la pupille de l'œil atteint de la tumeur prend généralement une couleur blanche ou rosâtre, souvent appelée leucocorie. Si elle n'est pas diagnostiquée et traitée, les conséquences peuvent être graves.
Qu'est-ce que le rétinoblastome ?
Le rétinoblastome est une tumeur maligne originaire de la rétine. Elle se développe généralement dans les zones sensibles à la lumière de la rétine, où elle convertit la lumière entrante en signaux visuels. Il s'agit de la tumeur maligne intraoculaire la plus courante de la petite enfance et se manifeste généralement au cours des trois premières années de vie.
Le rétinoblastome peut toucher un œil ou les deux yeux ; environ un quart des cas touchent les deux yeux. On pense qu'il est dû à des anomalies génétiques liées à la différenciation des cellules rétiniennes. Il est généralement, mais pas toujours, diagnostiqué avant l'âge de trois ans, bien que des cas surviennent chez l'adulte après une période de latence.
Types de rétinoblastome
Le rétinoblastome se présente sous trois formes principales :
- Rétinoblastome unilatéral : il ne touche qu'un seul œil.
- Rétinoblastome bilatéral : il touche les deux yeux.
- Rétinoblastome trilatéral : il s'agit d'une affection inhabituelle dans laquelle la tumeur se trouve à la fois dans les yeux et dans la glande pinéale du cerveau.
Environ 60 % de ces rétinoblastomes sont unilatéraux, tandis que les rétinoblastomes bilatéraux et trilatéraux représentent 40 %.
Quelle est la fréquence du rétinoblastome ?
Le rétinoblastome est considéré comme une maladie très rare ; seulement environ 3,3 personnes sur 1 million de moins de 20 ans sont diagnostiquées avec ce trouble. On estime que chaque année, environ 300 nouveaux cas sont découverts aux États-Unis, tandis qu'à l'échelle mondiale, on estime qu'environ 9 000 nouveaux cas sont diagnostiqués chaque année.
Symptômes et causes
Symptômes du rétinoblastome
Les symptômes de la maladie sont quelque peu difficiles à décrire du point de vue d'un enfant, en plus du fait que le diagnostic est généralement posé avant l'âge de trois ans. Par conséquent, d'autres signes, tels que des changements d'apparence ou de comportement, doivent être au centre des préoccupations des soignants.
Le symptôme le plus précoce et le plus fréquent qui se présente est la leucocorie, qui comprend une blancheur ou une pâleur de la pupille dans différentes conditions de lumière, en particulier sur les photographies au flash. D'autres symptômes qui pourraient impliquer la possibilité d'un rétinoblastome incluent :
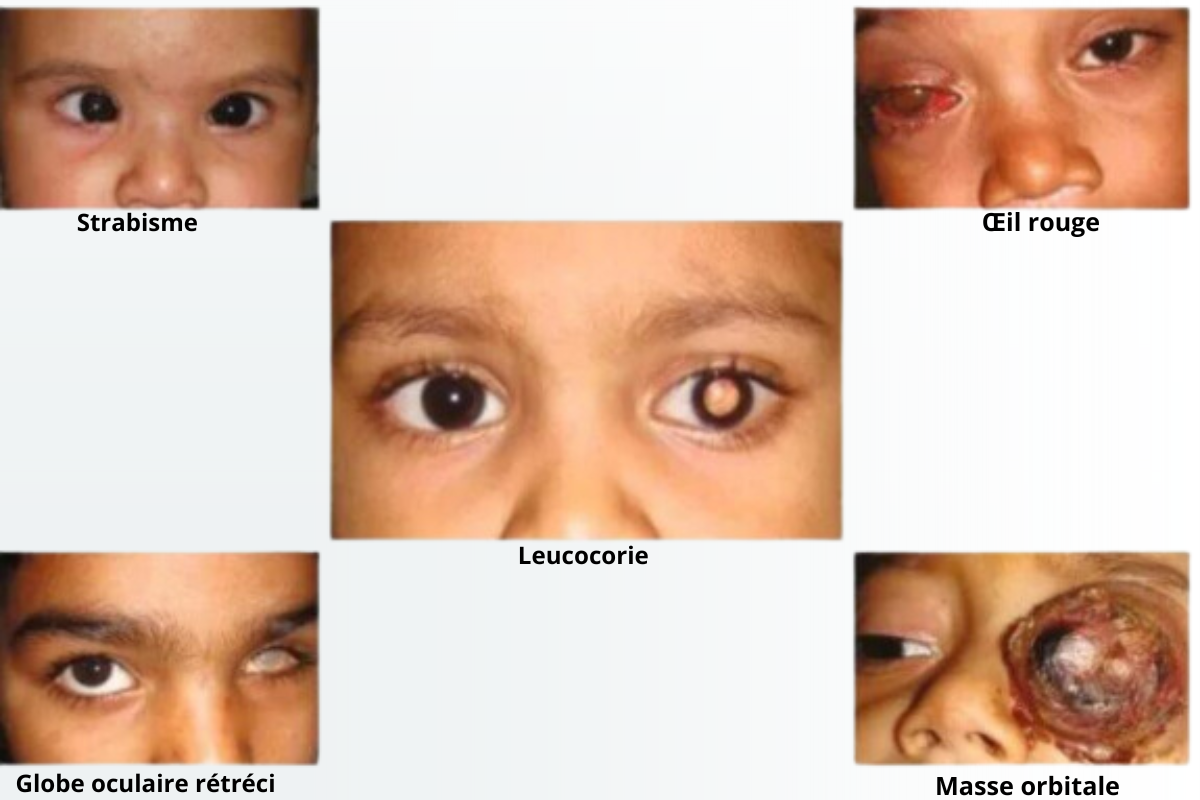
- Strabisme : yeux mal alignés qui ne se concentrent pas sur le même objet.
- Problèmes de vision : difficulté à suivre des objets en mouvement ou absence de mouvements oculaires.
- Douleur : irritabilité accrue, difficulté à dormir ou changement des habitudes alimentaires.
- Agrandissement de l'œil : la buphtalmie est une affection dans laquelle l'œil est plus gros que la normale.
- Yeux exorbités : également appelé proptose, une affection dans laquelle l'œil est exorbité hors de son orbite.
- Sang dans l'œil : l'hyphéma est une affection hémorragique de la chambre antérieure de l'œil.
- Infections oculaires : les symptômes comprennent une rougeur ou un gonflement dans ou autour de l'œil.
Causes du rétinoblastome
Le rétinoblastome survient lorsque les cellules de la rétine commencent à développer des changements cancéreux et à croître de manière incontrôlable. Cette croissance incontrôlable peut conduire à des tumeurs et peut métastaser ou se propager à d'autres zones du corps.
Ce défaut peut souvent être attribué à un problème dans l'ADN. Il est utile de considérer l'ADN comme une sorte de livre de recettes pour le comportement cellulaire. Les erreurs dans l'ADN peuvent entraîner un mauvais type de comportement cellulaire, comme une croissance incontrôlée.
Les tests et conseils génétiques sont recommandés pour tous les enfants diagnostiqués avec un rétinoblastome, que la mutation soit considérée comme héréditaire ou non. Ils sont également recommandés pour les frères et sœurs biologiques et les autres membres de la famille.
La mutation génétique associée à la maladie du rétinoblastome comprend le gène RB1, qui est un suppresseur de tumeur important. Sa fonction principale consiste à contrôler la croissance cellulaire. En raison d'un gène muté affecté, la capacité à exercer une régulation sur la reproduction et la croissance des cellules de la rétine peut être perdue ; par conséquent, le développement de la tumeur peut se produire. Dans certains cas, la délétion du chromosome 13p, qui héberge le gène RB1, a été mise en cause.
Le rétinoblastome peut être dû à deux principaux types d'erreurs génétiques :
- Cas sporadiques : il s'agit des cas dans lesquels une erreur se produit lors de la réplication de l'ADN, y compris dans un seul œil. Il s'agit d'une erreur de frappe lors de la copie d'une recette.
- Cas héréditaires : une mutation génétique est présente chez l'un ou les deux parents. L'enfant obtient la mutation et possède donc un gène avec environ 50 % de chances de développer la maladie si un seul des parents est affecté, et environ 75 % si les deux parents sont porteurs du gène. Chose choquante, certains porteurs ne présentent aucun symptôme de la maladie.
De plus, les frères et sœurs d'un enfant diagnostiqué avec un rétinoblastome ont un risque accru d'être atteints de la maladie même si les parents ne l'ont pas. Le risque pour un frère ou une sœur d'être également atteint d'un rétinoblastome est d'environ 4 à 7 %.
Complications du rétinoblastome
Les conséquences graves du rétinoblastome peuvent entraîner la destruction des tissus environnants, voire la cécité d'un ou des deux yeux. Le rétinoblastome peut métastaser, augmentant la gravité du cancer, ce qui nécessiterait des méthodes de traitement plus agressives. La voie de métastase par le nerf optique vers le cerveau, qui entraîne des tumeurs cérébrales secondaires, est particulièrement préoccupante.
De plus, les changements génétiques conduisant au rétinoblastome laissent une personne avec un risque d'autres tumeurs malignes plus tard dans sa vie. Les survivants auraient un risque annuel cumulé d'environ 1 %, qui passe à environ 20 % à l'âge de 20 ans. Les cancers secondaires les plus courants sont les suivants :
- Sarcomes : ils touchent les os et les tissus conjonctifs.
- Mélanomes : ils touchent la peau, les yeux et les muqueuses.
- Cancer du poumon : en raison de l'apport vasculaire riche dans les poumons, les tumeurs métastasent facilement dans d'autres parties du corps.
Diagnostic et tests
Comment diagnostique-t-on le rétinoblastome ?
Le diagnostic du rétinoblastome se fait généralement lorsque les parents ou les personnes qui s'occupent de l'enfant constatent une leucocorie et demandent conseil à un pédiatre. Le pédiatre peut confirmer la leucocorie lors d'un examen physique et oriente généralement l'enfant vers un ophtalmologue.
Un ophtalmologue examinera l'intérieur de l'œil, qui est généralement dilaté avec des gouttes médicamenteuses, et parfois l'examen se fait sous anesthésie pour les très jeunes enfants. Les examens d'imagerie sont importants dans le diagnostic du rétinoblastome et comprennent :
- Échographie : elle est utile pour localiser les dépôts de calcium, qui sont la marque habituelle de la plupart des rétinoblastomes.
- TDM : parfois très utile pour diagnostiquer les dépôts de calcium et délimiter la tumeur présente.
- Imagerie par résonance magnétique (IRM) : elle fournit des images détaillées de l'œil et des structures environnantes, qui sont importantes pour la stadification de la dissémination tumorale.
- Tomographie par émission de positons (TEP) : cet examen permet de déterminer si la tumeur s'est propagée à d'autres parties du corps.
Prise en charge et traitement
Comment traite-t-on le rétinoblastome ?
Le traitement du rétinoblastome dépend de la taille de la tumeur, de son emplacement et de sa propagation. Souvent, plusieurs traitements sont utilisés en combinaison et peuvent impliquer un ou plusieurs des éléments suivants :
- Chimiothérapie : il s'agit d'un traitement dans lequel des médicaments sont utilisés pour détruire les cellules cancéreuses. Dans le cas du rétinoblastome, la chimiothérapie est généralement administrée par voie intraveineuse et peut être un traitement néo-adjuvant pour réduire la taille de la tumeur avant d'opter pour une autre forme de traitement.
- Cryothérapie : il s'agit d'un traitement qui consiste à appliquer du froid sur la tumeur pour la détruire. La cryothérapie est généralement utilisée pour traiter les petites tumeurs, moins de 3 cm de large.
- Photothérapie dynamique (PDT) : il s'agit d'un traitement au laser qui cible la tumeur, mais nécessite souvent l'utilisation d'un médicament photo-sensibilisant.
- Radiothérapie : il s'agit d'un traitement qui utilise des rayons X de haute énergie pour cibler et détruire les cellules cancéreuses. Ce traitement est généralement utilisé pour traiter des tumeurs avancées et peut avoir des effets secondaires importants.
- Chirurgie : il s'agit d'une méthode qui consiste à retirer la tumeur ou l'œil atteint. L'enlèvement de l'œil est considéré comme la dernière option en raison de l'impact psychologique que cela peut avoir sur un enfant.
Conclusion
Le rétinoblastome est une maladie rare qui peut avoir des conséquences graves sur la vision et la vie d'un enfant. Un diagnostic précoce est essentiel pour améliorer le pronostic et la prise en charge. Les soins médicaux appropriés sont cruciaux pour la gestion de la maladie, et un suivi régulier est indispensable pour surveiller la santé de l'enfant tout au long de sa vie.
