Retinoblastoma is a rare but potentially deadly cancer of the retina, which is a light-sensitive tissue in the rear of the eye. One of the most noticeable signs of this condition is that the pupil of the eye with the tumour typically takes on a white or pinkish colour, often called leukocoria. If left undiagnosed and untreated, the outcome can be severe.
What is Retinoblastoma?
Retinoblastoma is a malignant tumour originating from the retina. It usually occurs in the light-sensitive areas of the retina, where it converts the incoming light into visual signals. It is the most common intraocular malignancy of early childhood and usually presents in the first three years of life.
Retinoblastoma can affect one eye or both eyes; roughly one-fourth of cases involve both eyes. It is believed to occur due to genetic abnormalities related to the differentiation of retinal cells. It usually, but not always, is diagnosed before age three, though instances of it arising in adults following a period of latency do occur.
Types of Retinoblastoma
Retinoblastoma has three main types:
- Unilateral Retinoblastoma: This involves only one eye.
- Bilateral Retinoblastoma: This involves a tumour in both eyes.
- Trilateral Retinoblastoma: This is an unusual condition where the tumour is in both the eyes and also in the pineal gland of the brain.
About 60% of these retinoblastomas are unilateral, while bilateral and trilateral are 40%.
How Common is Retinoblastoma?
Retinoblastoma is thought to be a very uncommon disease; only about 3.3 per 1 million individuals below the age of 20 years are diagnosed with this disorder. It is estimated that every year, around 300 new cases are discovered in the United States, while worldwide it is estimated that approximately 9,000 new cases are diagnosed annually.
Symptoms and Causes
Symptoms of Retinoblastoma
The symptoms of the disease are somehow hard to describe from a child's perspective besides; it is generally diagnosed below the age of three. Therefore, other signs, such as appearance changes or behaviors should be the point of concern for the caregivers.
The earliest and most frequent symptom that presents itself is leukocoria, which includes whiteness or paleness of the pupil under different light conditions, especially in flash photographs. Other symptoms that could imply the possibility of having retinoblastoma:
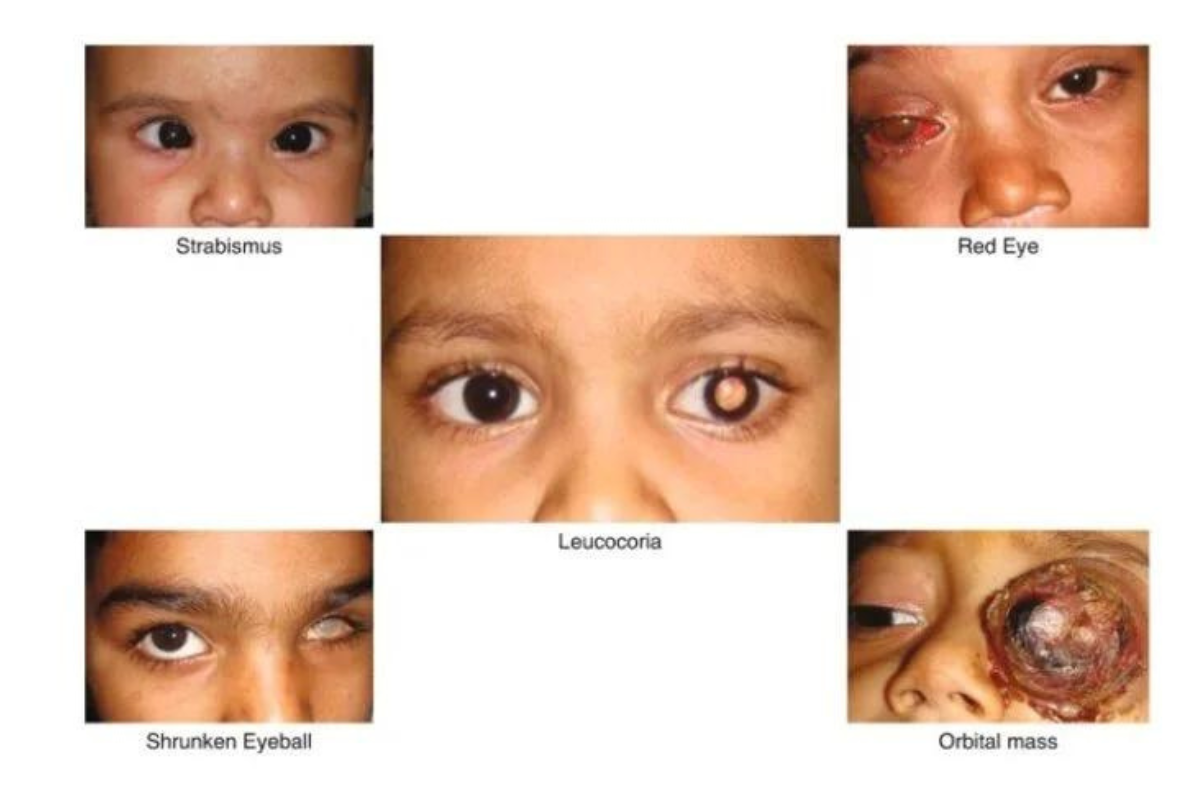
- Strabismus: Misaligned eyes that do not focus on the same object. Vision Problems: Difficulty in following objects that are moving, or the lack of eye movements. Pain: Increased irritability, difficulty sleeping, or a change in feeding habits. Eye
- Enlargement: Buphthalmos is a condition where the eye is bigger in appearance than normal. Bulging Eyes: This is also called proptosis, a condition where the eye is bulging out of its orbit.
- Blood in the Eye: Hyphema is a bleeding condition of the front chamber of the eye.
- Eye Infections: Symptoms include redness or swelling in or around the eye.
Causes of Retinoblastoma
Retinoblastoma occurs when the cells of the retina start to develop cancerous changes and grow uncontrollably. This uncontrollable growth can lead to tumors and can metastasize, or spread to other areas of the body.
This defect can often be traced to a problem in the DNA. It's useful to consider DNA as a kind of cookbook with recipes for cellular behaviour. Mistakes in DNA can result in the wrong kind of cellular behaviour, such as uncontrolled growth.
Genetic testing and counselling are recommended for all children diagnosed with retinoblastoma, whether or not the mutation is considered inherited. It is also recommended for biological siblings and other family members.
The genetic mutation associated with the disease retinoblastoma includes the RB1 gene, which is an important tumour suppressor. Its principal function consists of controlling cell growth. Due to an affected mutated gene, the capacity to exert regulation on the reproduction and growth of the cells within the retina might be lost; hence, tumour development may happen. In some cases, deletion of chromosome 13p, hosting the RB1 gene, has been implicated.
Retinoblastoma may occur due to two major types of genetic mistakes:
Sporadic cases: These are the cases in which some mistake occurs when DNA is replicating, including only one eye. It is a typo while copying a recipe.
Inherited Cases: A genetic mutation is present in one or both parents. The child obtains the mutation and hence has a gene with about a 50% chance of developing the condition if only one of the parents is affected, and about 75% if both parents are carriers of the gene. Shockingly, some carriers do not exhibit symptoms of the disease.
Also, the siblings of a child diagnosed with retinoblastoma are at an increased chance of having the disease even though the parents may not have it. The risk for a sibling to also have retinoblastoma is around 4-7%.
Complications of Retinoblastoma
The serious consequences of retinoblastoma include the destruction of the surrounding tissues, and even blindness in one or both eyes. Retinoblastoma may metastasize, increasing the severity of the cancer which would necessitate more aggressive treatment methods. Of most concern is the metastasis route through the optic nerve into the brain, resulting in secondary brain tumours.
Furthermore, the genetic changes leading to retinoblastoma leave a person with a risk of other malignancies later in their life. The survivors are reported to be at a cumulative annual risk of about 1%, which rises to about 20% by the age of 20 years. The commonest secondary cancers include:
- Sarcomas: These involve bones and connective tissues.
- Melanomas: These are related to skin, eyes, and mucous membranes.
- Lung cancer: Due to the rich vascular supply in the lung, tumours easily metastasis to other parts of the body
Diagnosis and Tests
How is Retinoblastoma Diagnosed?
Diagnosing retinoblastoma usually occurs when leukocoria is observed by parents or caretakers who seek advice from a paediatrician. The paediatrician may confirm leukocoria during a physical examination and normally refers the child to an ophthalmologist.
An ophthalmologist will look into the interior of the eye, which is usually dilated with medicated drops, and at times the examination is under anaesthesia for very young children. Imaging studies are important in the diagnosis of retinoblastoma and include:
- Ultrasound: This is useful for locating calcium deposits, which are the usual trademark of most retinoblastomas.
- CAT Scan: Sometimes very helpful in diagnosing calcium deposits and outlining the tumour present.
- Imaging Studies Magnetic Resonance Imaging (MRI) Scan: These provide detailed pictures of the eye and its surrounding structures, which are significant in the staging of tumour dissemination.
- Positron Emission Tomography (PET) Scan: This test can determine if the tumour has spread to other body parts.
Management and Treatment How is Retinoblastoma Treated?
Treatment for retinoblastoma depends on the size of the tumour, its location, and whether the tumour has spread. Often, several treatments are used in combination and may involve one or more of the following:
- Chemotherapy: This is a treatment where drugs are given that destroy the cancerous cells. It can be systemic, through IV infusion, or local, by injections to target a region. In this setting, chemotherapy has the aim of shrinking the tumours to preserve vision with limited surgical intervention.
- Radiation Therapy: High-energy radiation is used to destroy tumour cells. However, radiation is mostly kept at a minimum due to its potential to cause complications in the long run.
- Focal Therapies: Cryotherapy, freezing treatment, thermotherapy, heat treatment, and laser therapy are methods that attack tumour cells directly.
- Surgery: Surgery may be required to stop the spread of the disease; this might involve removing the affected eye in cases where the tumour is very large. It is usually reserved for those situations where useful vision is already lost.
- Supportive Care: This may include medications to address such side effects as nausea and vomiting, which frequently accompany chemotherapy.
Prevention
Is Retinoblastoma Preventable?
As genetic alterations represent the major cause of retinoblastoma, the disease itself cannot be prevented. Those individuals with a family medical history regarding this disorder, or those with known gene mutations, may avail themselves of genetic counselling to understand the risk of passing on the disease to offspring.
Outlook / Prognosis
What Can I Expect if I Have (or My Child Has) This Condition?
The prognosis of retinoblastoma, in the majority of instances, is good, with an overall survival rate of about 95% in pediatric cases. The likelihood of a favourable outcome, including that of sight preservation, is very high if diagnosed before the age of two.
Survivors of retinoblastoma will require lifelong follow-up for the development of new cancers, often consisting of periodic scans to allow for the early detection of any new tumours. Healthcare providers should be consulted for recommendations on what follow-up care is needed.
Living With Retinoblastoma
When Should I Call My Healthcare Provider?
If the above signs and symptoms suggest that a child may be suffering from retinoblastoma, or if any change in the vision of a child is noticed, then the child should be taken to a doctor for consultation. If there is a family medical history of Retinoblastoma, or if mutations in the RB1 gene are known, then one should share their apprehensions with a doctor.
History of Retinoblastoma in the Family
It is necessary for all families with a history of developing retinoblastoma to go for periodic checkups of their children and other family members. The RB1 gene may lead to the development of not only retinoblastoma but also a benign retinal tumour called retinocytoma. For a child to have an excellent outcome, it is very important to be treated and diagnosed early through periodic screening. This is very crucial.
Conclusion
In all, retinoblastoma is an extremely rare but serious condition that can have a profound impact on children and their families. Early diagnosis and a multidisciplinary approach to treatment can result in a much better survival rate and quality of life. This will enable families, through better awareness of symptoms and follow-up checks, to cooperate with healthcare providers in managing the condition.
